A new AI-based system called SIGNET maps causal gene regulatory networks across six brain cell types in Alzheimer's brains, revealing extensive gene rewiring—especially in excitatory neurons—and identifying hub genes that could serve as early diagnostic markers or therapeutic targets.
Using single-cell RNA sequencing on cysts from mice, researchers found that brain cysts of Toxoplasma gondii contain multiple bradyzoite subtypes rather than a single dormant form. These subtypes support survival, spread, or reactivation, explaining why current drugs can’t eradicate cysts and suggesting new therapy targets; the finding reshapes the parasite’s life cycle and underscores congenital toxoplasmosis risk worldwide.
A UC Riverside study using single-cell RNA sequencing finds that Toxoplasma gondii cysts in mouse brains contain multiple parasite subtypes, not just dormant forms, with some subtypes showing growth and reactivation potential, challenging the idea of a single dormant cyst and highlighting new targets for toxoplasmosis treatments.
A 12-week trial led by Dr. Wooje Lee at the World Institute of Kimchi found that daily kimchi powder can subtly tune the immune system, boosting antigen presentation and regulated CD4+ T-cell activation without causing broad immune stimulation. Effects were observed in overweight adults using single-cell RNA sequencing, with fermentation method (traditional vs. starter-culture Leuconostoc mesenteroides) influencing strength of signals. B cells and cytotoxic T cells were largely unchanged, and illness outcomes were not measured. Further international research is planned.
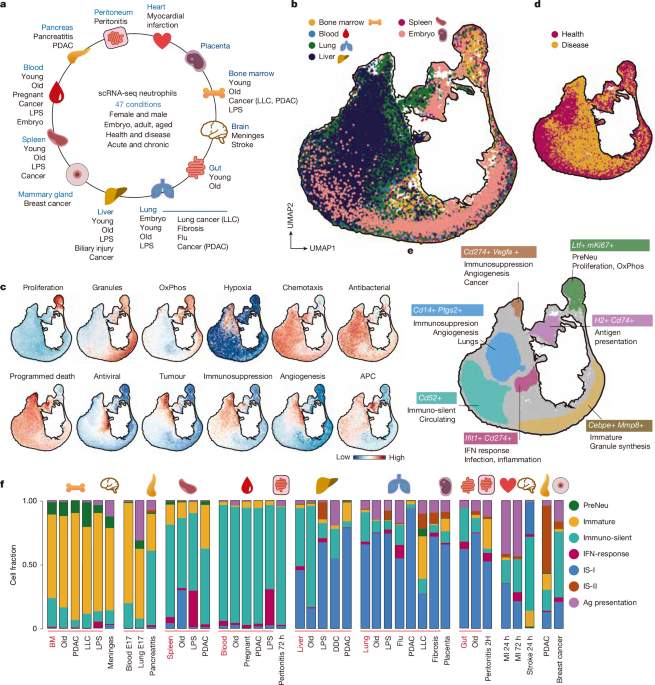
The article presents NeuMap, a comprehensive transcriptional map of neutrophil diversity across tissues, developmental stages, and disease conditions in mice and humans, revealing a limited set of conserved functional states or hubs that are dynamically regulated by signals and transcription factors, with implications for understanding neutrophil roles in health and disease.
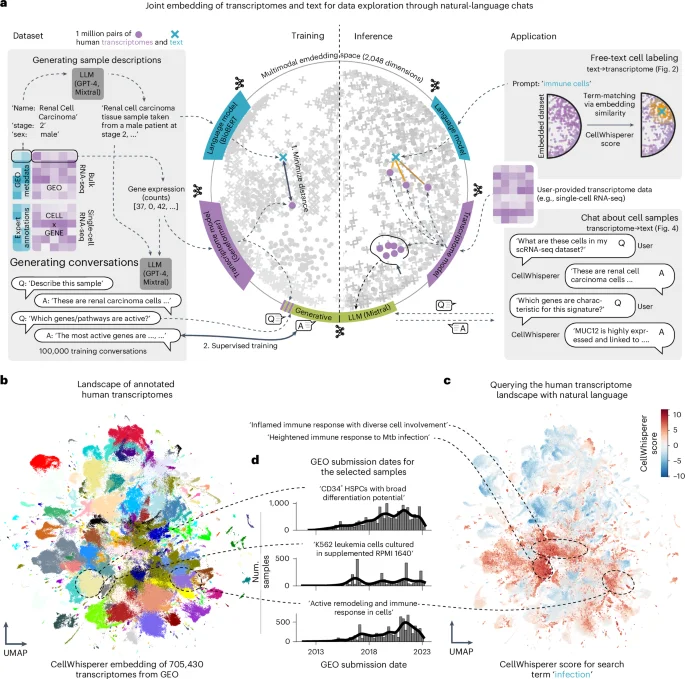
The article introduces CellWhisperer, a multimodal AI framework that enables chat-based exploration and analysis of single-cell RNA-seq data using natural language, integrating transcriptome profiles with biological text understanding to facilitate intuitive data interrogation and hypothesis generation.
The study investigates how limited β-cell death influences pancreatic islet macrophages, revealing efferocytic remodeling and similarities with pathology-associated macrophages in other tissues, which may impact autoimmune diabetes development.
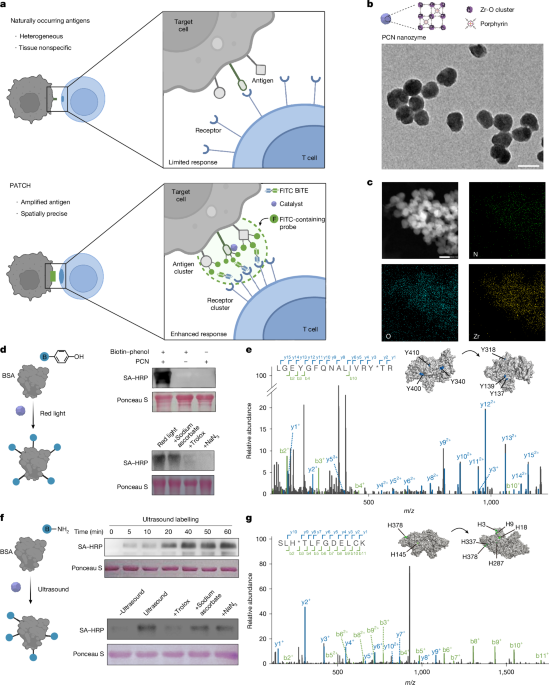
The article discusses a study on enhancing antigen-induced cellular responses using proximity labelling techniques, with data available from various repositories and analysis code on GitHub, highlighting advances in immune signaling and proteomics methods.
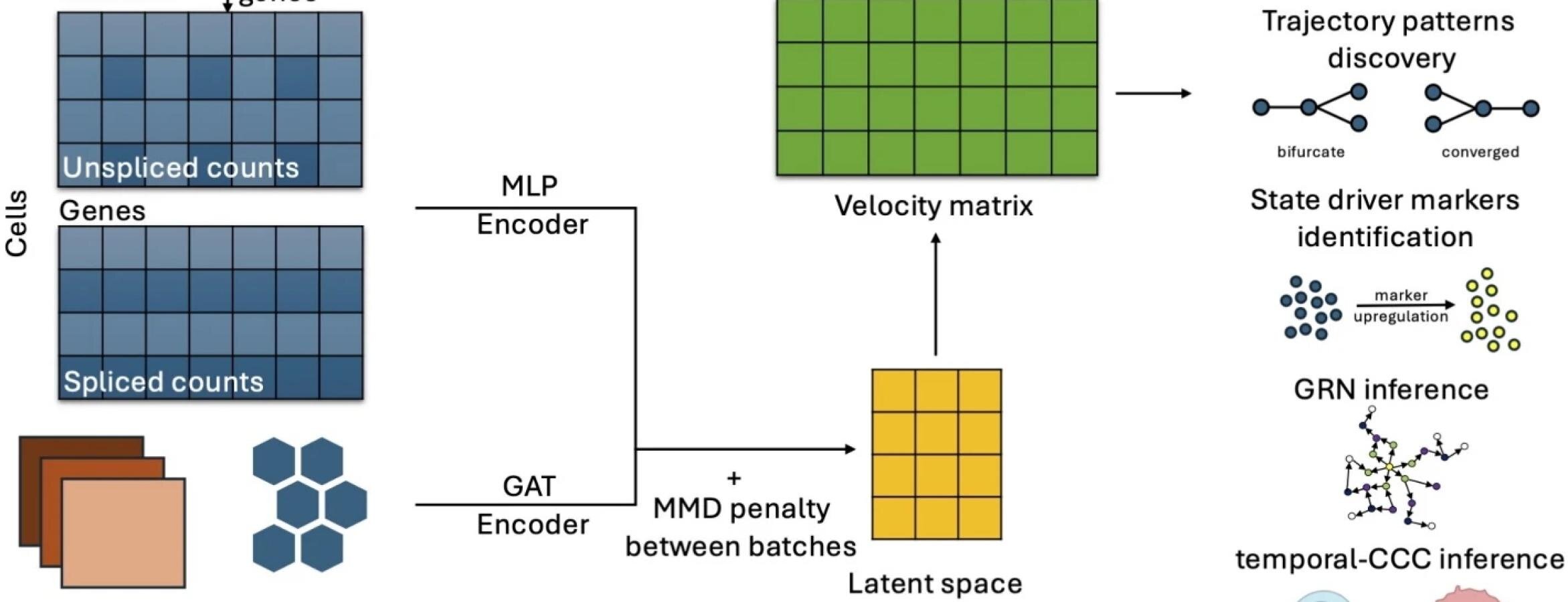
Researchers developed spVelo, a new method using neural networks to improve the measurement of gene expression changes and cell fate decisions by incorporating spatial and batch information, enabling more accurate and comprehensive analysis of cellular development and differentiation.
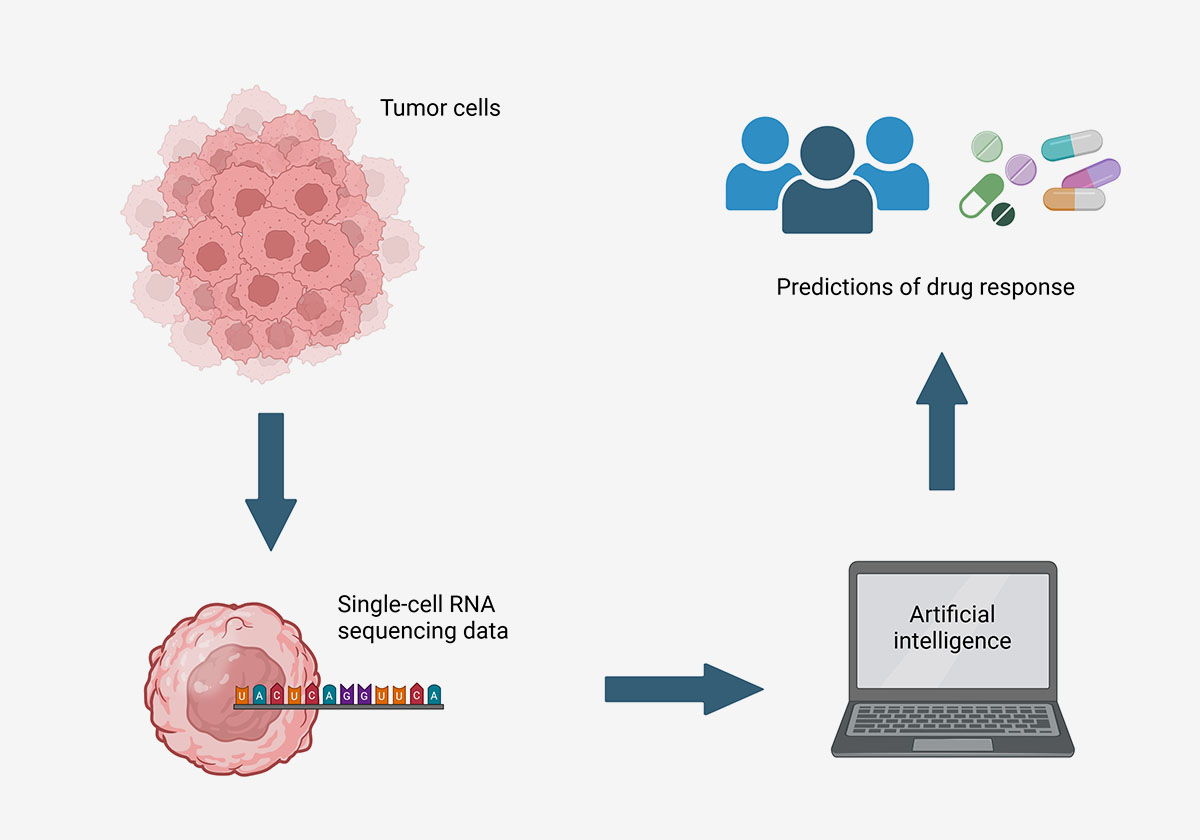
Researchers at the National Institutes of Health (NIH) have developed an AI tool that uses single-cell RNA sequencing data to predict whether a person’s cancer will respond to a specific drug, potentially leading to more precise matching of cancer patients with effective drugs. The tool, called Personalized Single-Cell Expression-based Planning for Treatments In Oncology (PERCEPTION), was able to accurately predict drug responses at the individual cell level and identify resistance to certain drugs in cancer patients. The researchers believe that this approach could lead to more lasting drug responses, but caution that its accuracy will improve as single-cell RNA sequencing data become more widely available.
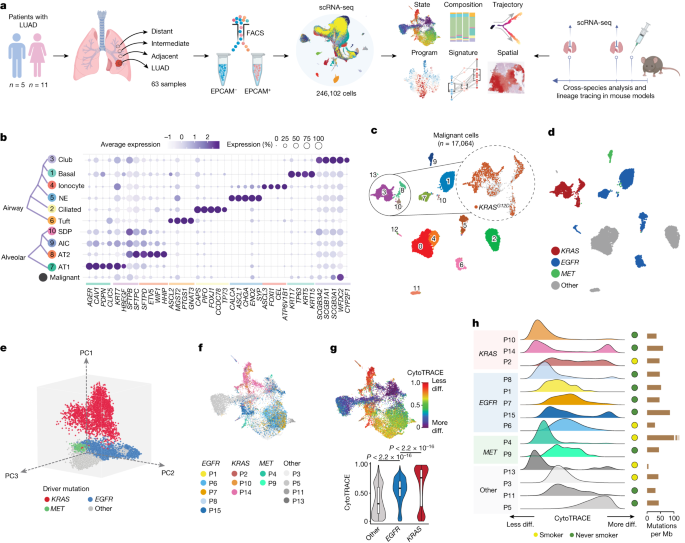
A study on lung adenocarcinoma (LUAD) reveals the transcriptional landscape of epithelial and malignant cells, identifying distinct cell subsets and their roles in LUAD development. The research shows that KRAS mutant LUADs exhibit unique transcriptional programs and extensive heterogeneity, while a subset of alveolar intermediate cells (AICs) transition to malignant cells, particularly KRAS mutant cells. These AICs, termed KRT8-high alveolar cells (KACs), are enriched in tumour-adjacent normal regions and display features of reactive epithelial cells. The study provides insights into the cellular and transcriptional phenotypes of LUAD, potentially identifying actionable targets for early treatment strategies.
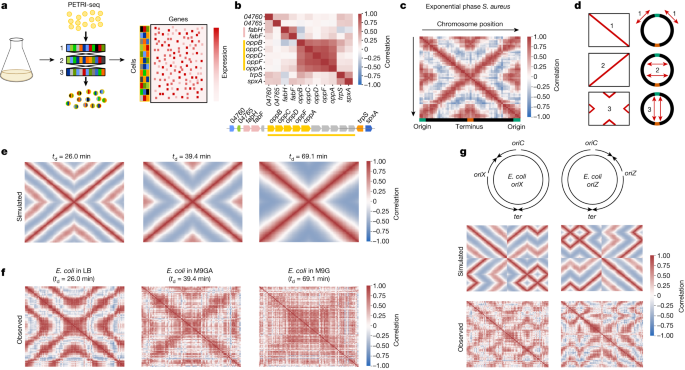
A study on transcription-replication interactions in bacterial genomes has been conducted, revealing insights into bacterial gene regulation mechanisms. The research utilized single-cell RNA sequencing analysis and bulk analysis of C. crescentus expression, providing valuable data available in public repositories. The study sheds light on the complex regulatory networks governing bacterial cell cycles and gene expression, offering potential applications in synthetic biology.
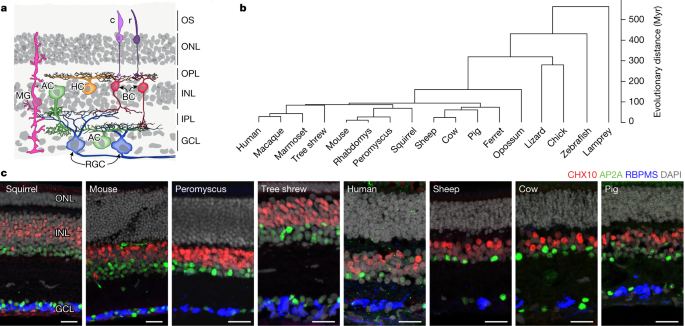
Researchers have used single-cell transcriptomics to compare retinal cell classes, subclasses, and types in 17 vertebrate species. They found that the functional and morphological characteristics of retinal cell classes are conserved across species, with marked similarities in gene expression. The study also revealed the evolutionary variation among cell types within photoreceptors, horizontal cells, bipolar cells, and retinal ganglion cells (RGCs). While many types were conserved, RGCs showed more extensive variation, suggesting that natural selection plays a role in shaping the retinal output. The identification of non-primate orthologues of midget RGCs, responsible for high-acuity vision, suggests that these cell types evolved from ancestral cell types present in the common mammalian ancestor.
Researchers at UC San Diego have discovered a biomarker using single-cell RNA sequencing that can predict whether neurons will regenerate after injury. This finding could lead to the development of regenerative therapies for spinal cord injuries and other neurological conditions. The biomarker, called the Regeneration Classifier, was found to be consistently reliable in neurons across the nervous system and at different developmental stages. While the classifier is currently a tool for lab research, the researchers hope to use it to predict the effectiveness of regenerative therapies in preclinical contexts and move closer to clinical trials.
Researchers have discovered a new biomarker, called the "Regeneration Classifier," that can predict the likelihood of neuronal regeneration after injuries. Using single-cell RNA sequencing, the study focused on neurons in the corticospinal tract, which are crucial for movement control but have low regeneration potential. The biomarker was found to be reliable across different developmental stages and regions of the nervous system. While clinical applications are still distant, this discovery could pave the way for advanced treatments in nerve repair and regenerative therapies.