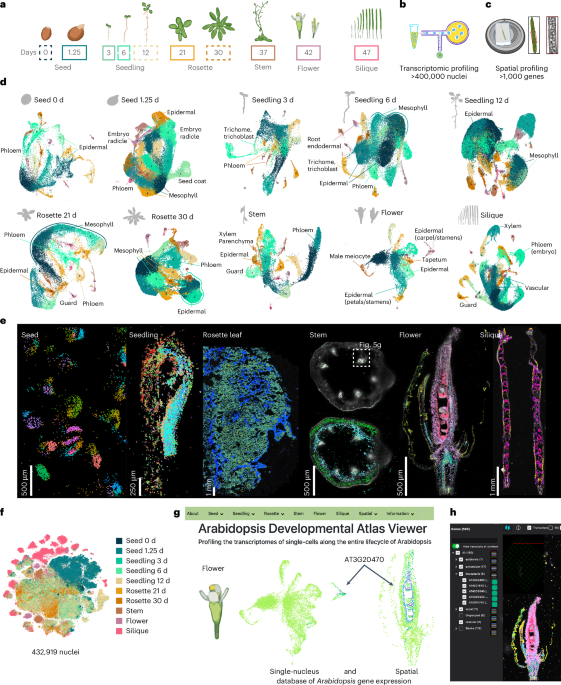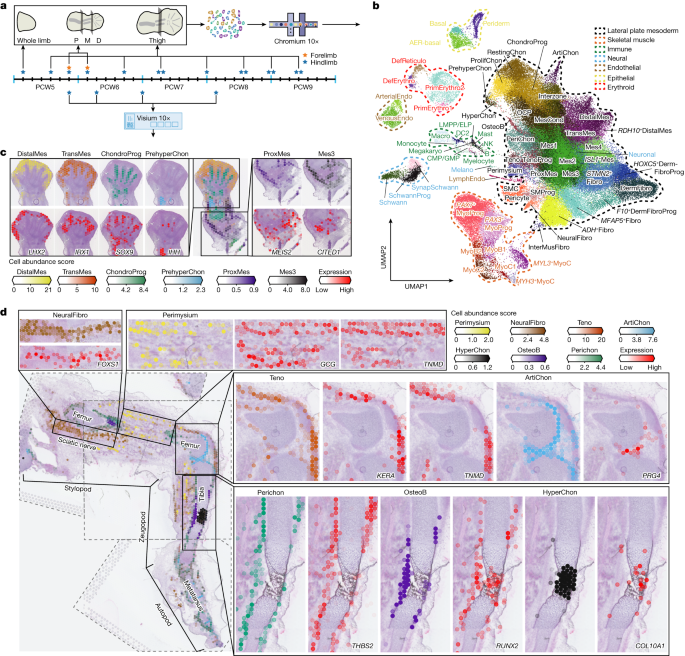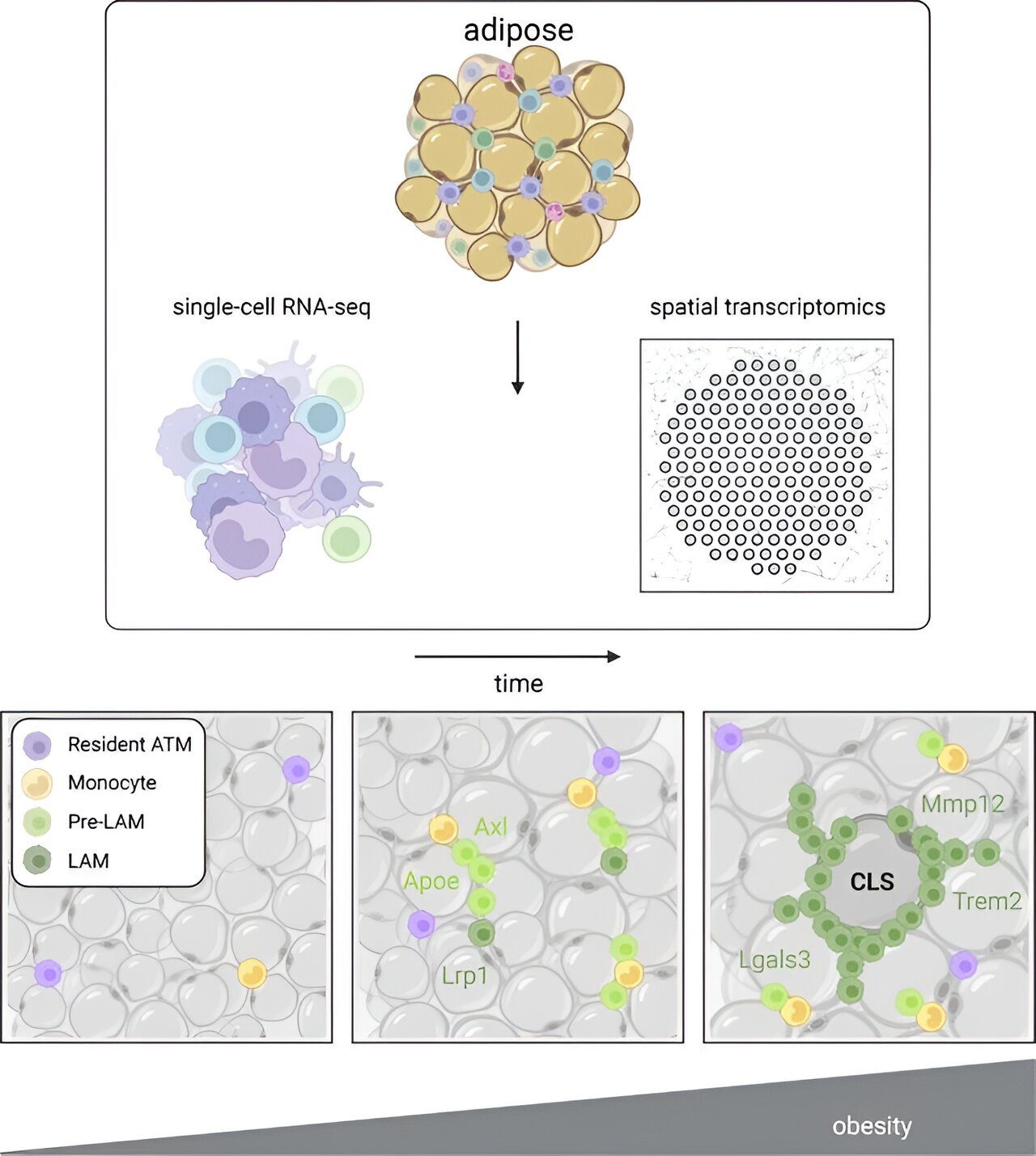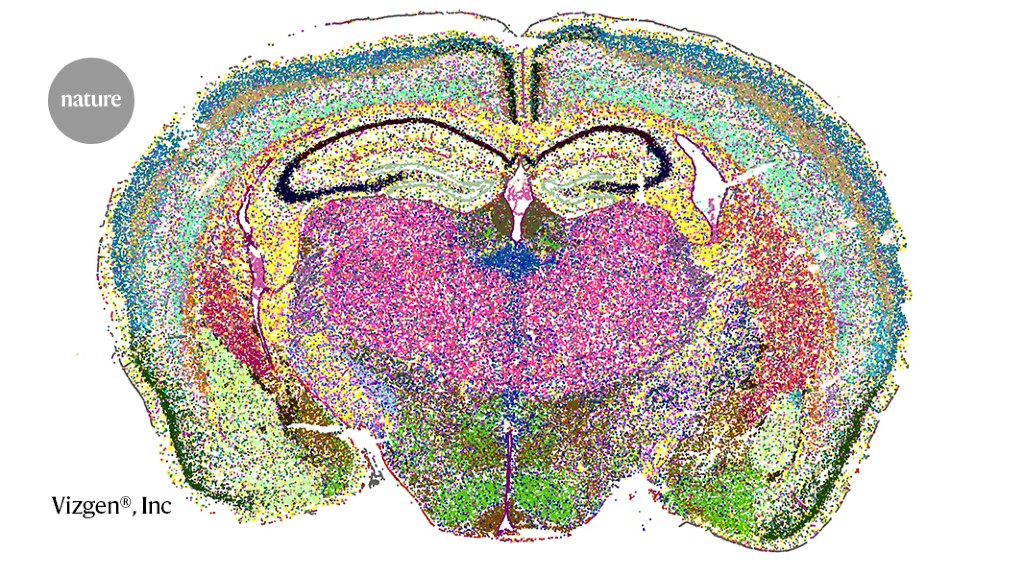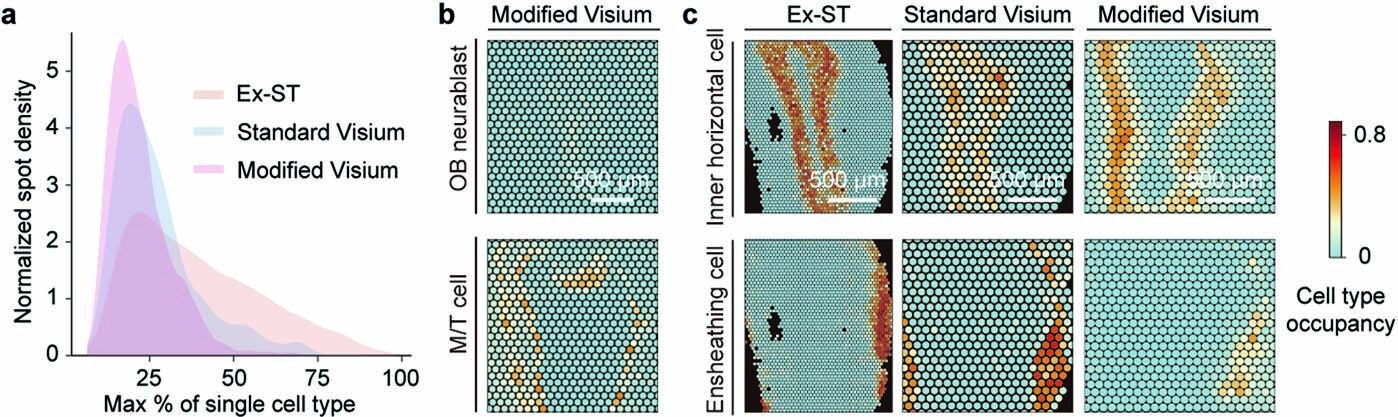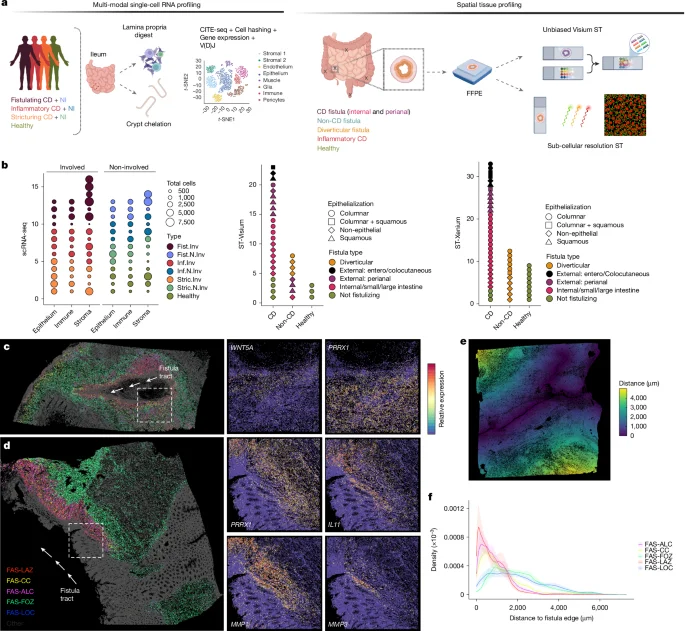
Fibroblast Niches Drive Crohn’s Fistula Formation
This study uses spatial transcriptomics and single-cell analysis to uncover how specialized fibroblast niches, particularly FAS cells, contribute to the formation, progression, and persistence of Crohn's fistulae, highlighting their roles in tissue remodeling, immune regulation, and epithelial regeneration, with implications for targeted therapies.