A new trimodal protein language model enhances the accuracy and efficiency of protein searches, leveraging advanced deep learning techniques to improve understanding of protein structures and functions.
Researchers used the advanced AI model AlphaFold3 to predict the structures of all 25 human bitter taste receptors, finding it more accurate than previous models. The study revealed conserved intracellular regions and variable extracellular regions, suggesting these receptors play a dual role in taste and gut-brain signaling, with implications for appetite control, glucose metabolism, and diabetes research.
Researchers at IBM and Moderna used a quantum computer to simulate the secondary structure of a 60-nucleotide-long mRNA sequence, the longest ever achieved with quantum simulation, demonstrating potential for more accurate vaccine development and overcoming limitations of classical and AI methods.
Scientists have discovered that disordered gene-regulating proteins rely on a structured partner, beta-catenin, to function properly, challenging previous ideas about their chaotic nature and revealing a hidden organization that could impact understanding of gene control and disease.
DeepMind has introduced AlphaFold3, an advanced AI-powered software that predicts the structure and function of every protein, significantly aiding scientific research in areas like drug discovery and bio-renewable materials. Unlike its predecessors, AlphaFold3 is not open source but is accessible for non-commercial research through the AlphaFold Server.
Scientists have decoded the molecular structure of a protein in the marine bristleworm Platynereis dumerilii that helps it sync its reproductive cycle with the phases of the moon. This is the first time the molecular structure of a protein responsible for syncing a biological clock to the moon has been determined. The protein, called L-Cry, can distinguish between moonlight and sunlight, allowing the worms to avoid mistaking the dim light of dawn and dusk for moonlight. The findings could have implications for understanding the physiology of other organisms, including humans, and shed light on the evolutionary origins of monthly reproductive timing.
Researchers have developed a method called AF-Cluster that uses sequence clustering to predict multiple conformations of proteins using AlphaFold2. This method was used to investigate the evolutionary distribution of predicted structures for the metamorphic protein KaiB and confirmed a surprising prediction about a cyanobacteria KaiB variant. The sensitivity of AF-Cluster to point mutations was also tested, and a putative alternate state was identified for the oxidoreductase Mpt53 in M. tuberculosis. This bioinformatic approach, in combination with experimental validation, has the potential to greatly impact the prediction of protein energy landscapes and understanding biological function.
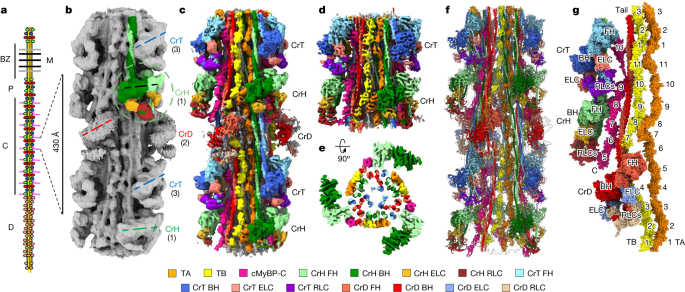
Researchers have used cryo-electron microscopy (cryo-EM) to determine the structure of the human cardiac myosin filament, a key component of muscle contraction in the heart. The study provides insights into the organization and arrangement of proteins within the filament, including myosin heads, tails, titins, and cMyBP-C. The structural data has been deposited in public databases, allowing other scientists to access and analyze the findings. This research contributes to our understanding of the molecular mechanisms underlying heart function and may have implications for the development of treatments for cardiac diseases.
Researchers are harnessing the power of artificial intelligence (AI) tools, such as AlphaFold and large language models, to prepare for future pandemics. These AI tools are being used to map the structure of viral proteins, identify mutations for vaccine development, predict virus evolution, and design new proteins for vaccine candidates. The use of AI in pandemic preparedness has shown promising results, but it is still in the early stages. Machine learning has the potential to accelerate vaccine design and stay ahead of evolving viruses, but caution is needed in relying solely on computational design.
The 2023 Lasker Awards were given to scientists who made significant advancements in the diagnosis of eye diseases and the prediction of cellular protein structure. James G. Fujimoto of MIT was awarded for his contribution to the invention of optical coherence tomography (OCT), an eye-scanning technology that can detect eye diseases earlier than previous methods, potentially preventing blindness. Additionally, the London-based AI lab DeepMind received an award for their work on protein folding, which has revolutionized the understanding of diseases and drug discovery. Dr. Piet Borst was recognized for his discoveries about parasitic diseases and cancer cell resistance to chemotherapy.
DeepMind's AlphaFold AI, which accurately predicts protein structures, has had a significant impact on the field of biology and drug discovery. The AlphaFold Protein Structure Database has been used by over 1.2 million researchers worldwide, and adoption rates are increasing rapidly. DeepMind CEO Demis Hassabis believes that AlphaFold has had the most beneficial effects in AI so far, with numerous Nobel Prize-winning biologists and chemists utilizing the technology. While AlphaFold has already made important contributions, the team acknowledges that there are still many unsolved problems in protein research. DeepMind continues to invest in AlphaFold and aims to see more real-world applications in the coming years.
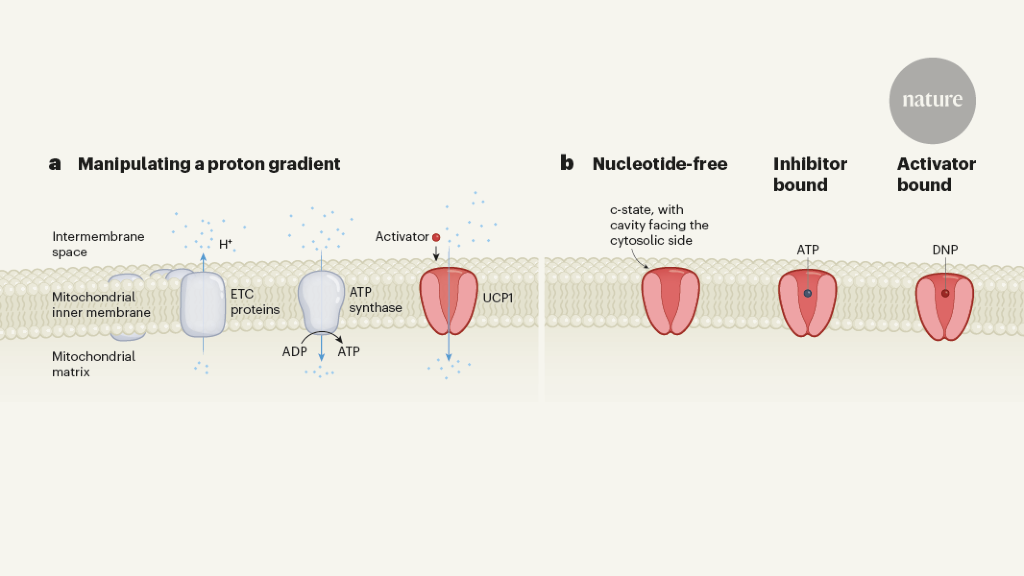
The structure of the human protein UCP1, which plays a role in releasing energy as heat in brown fat, has been obtained, providing valuable insights into its mechanism of action. This breakthrough could potentially aid in the development of drugs for obesity and other metabolism-related disorders.
Scientists have discovered the molecular structure of a protein called UCP1, which enables brown fat tissue to burn calories as heat. This breakthrough could lead to the development of treatments that activate UCP1 to burn off excess calories from fat and sugar, potentially combating obesity and diabetes. The research provides crucial details for the development of therapeutics that target UCP1 and could help increase brown fat activity to burn more calories and fight metabolic diseases.
Scientists have discovered the structure of the protein that triggers Huntington's disease, which could lead to a treatment that stops the disease in its tracks. The protein, called huntingtin, develops an abnormally long strand of repeating amino acids within its structure, called the polyglutamine stretch, which builds up in the brain and folds itself into a shape that is toxic to cells. The research team determined the structure of the amyloid nucleus for HTT protein, the "spark" that sets off the chain reaction of protein misfolding, and found that 36 polyQ repeats are the critical number for nucleation to happen in single protein molecules. Armed with this knowledge, the team hopes future work can explore ways of preventing nucleation so that the toxic cascade of protein misfolding is never triggered in the first place.
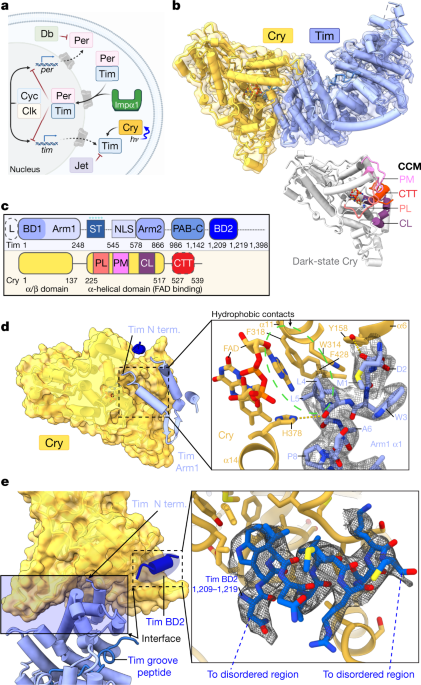
Researchers have revealed the structure of cryptochrome-timeless, a protein complex that plays a key role in the circadian clock. The study sheds light on the mechanisms behind the timing of the body's internal clock and could lead to new treatments for sleep disorders. The researchers used a combination of X-ray crystallography and cryo-electron microscopy to determine the structure of the complex, which is found in both humans and fruit flies.