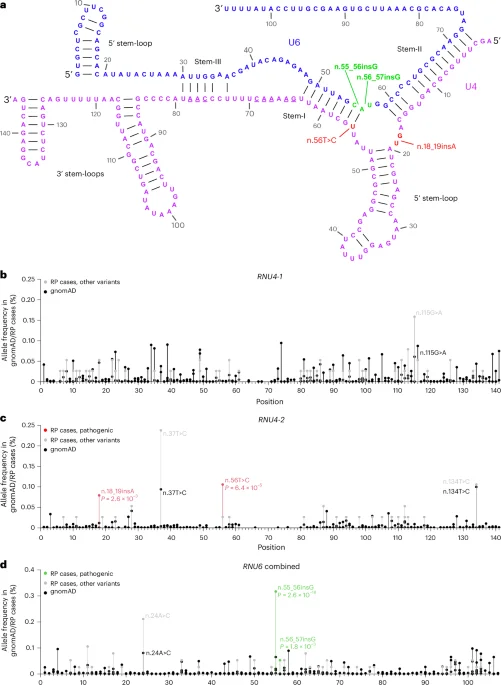
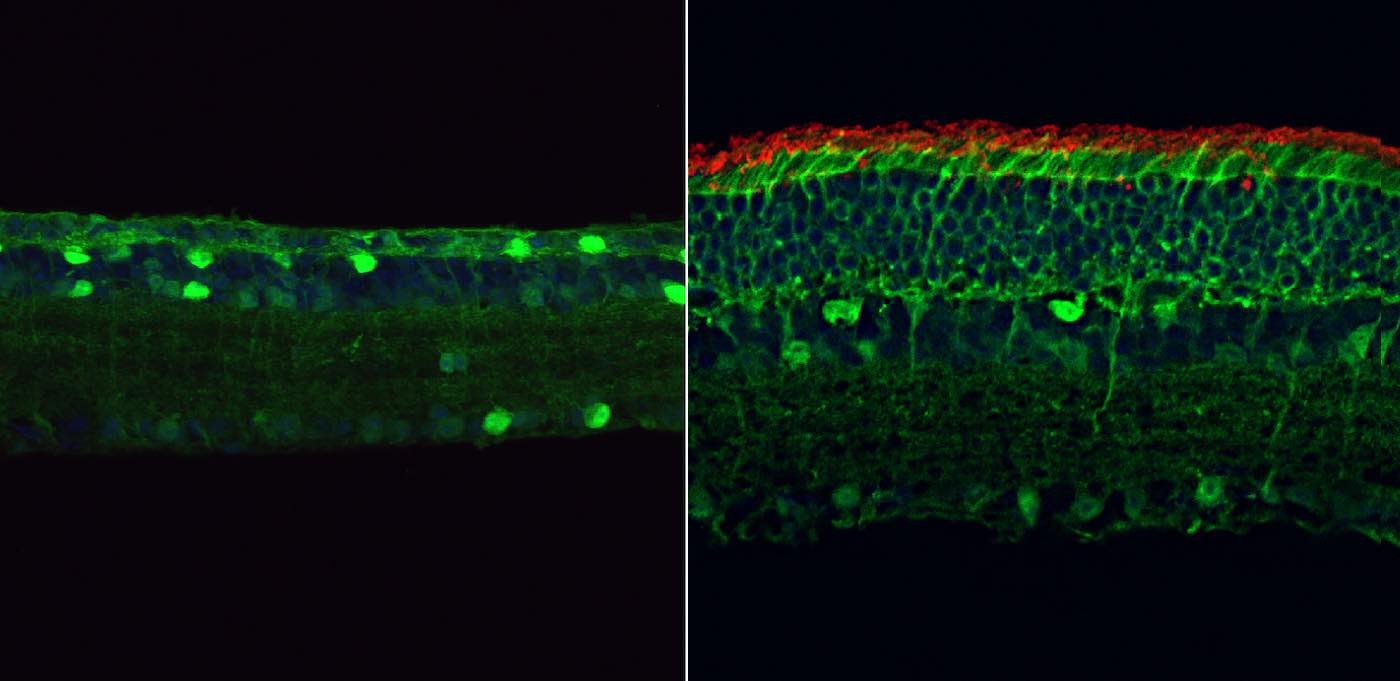
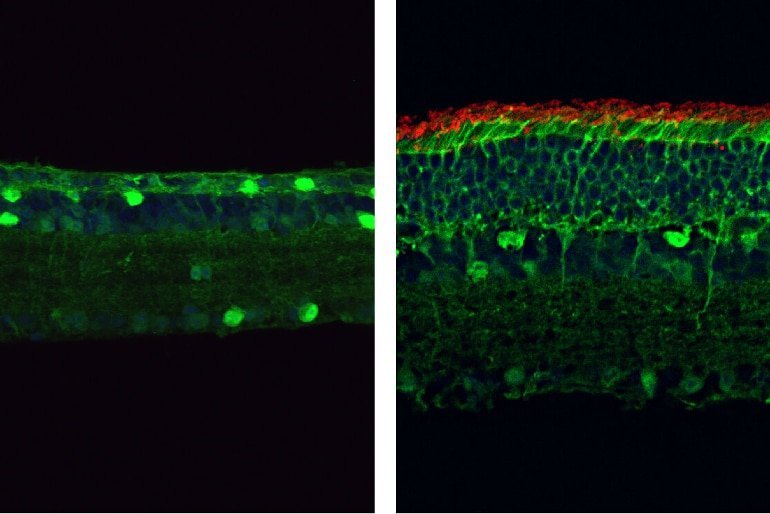
The study identifies de novo and inherited variants in U4 and U6 snRNA genes as causes of autosomal dominant retinitis pigmentosa, expanding the genetic landscape of the disease and highlighting the role of noncoding RNA mutations in retinal degeneration.
Scientists discovered that in retinitis pigmentosa, the retina can adapt by rerouting signals from cones to rod bipolar cells, helping preserve daylight vision longer despite rod loss, which could inform future treatments.
Research shows that retinal neurons can rewire themselves to maintain vision in retinitis pigmentosa, with rod bipolar cells forming new connections with cone cells as rods degenerate, a process triggered by degeneration itself and not just light response loss, offering potential targets for therapies to slow vision loss.
Researchers have developed solar-powered eye implants that could potentially help people with incurable eye diseases such as retinitis pigmentosa and age-related macular degeneration. The technology aims to power sensors and cameras in the eye using solar photovoltaic panels, eliminating the need for external charging. While still in the proof-of-concept stage, the idea shows promise in addressing the challenge of powering the implants and could significantly impact the treatment of eye diseases in the future.
Scientists from the University of California, Irvine, have discovered a nanobody that may lead to a treatment for Retinitis Pigmentosa (RP), a group of inherited eye diseases that cause vision loss. The nanobody targets the Rhodopsin molecule, a key light-sensing molecule in the retina, and can halt its photoactivation process. The nanobodies have shown high specificity and can recognize the target Rhodopsin extracellularly, potentially allowing for the development of gene therapies for RP. The researchers aim to improve the nanobodies' ability to recognize Rhodopsin from other species and resolve the key intermediate states of Rhodopsin for further study.
Researchers have discovered small-molecule drugs called stress resilience-enhancing drugs (SREDs) that could be used to treat age-related macular degeneration, diabetic retinopathy, and retinitis pigmentosa. The drugs slowed or halted the progression of retinopathies in animal models by inhibiting cyclic nucleotide phosphodiesterases. These chronic, progressive retinal diseases arise from genetic and environmental disruptions of cellular and tissue stability, and currently have limited treatment options. The researchers innovated a systems pharmacology platform that leverages state-of-the-art disease modeling and characterization to identify novel, mechanism-based therapies that mitigate disease at the root cause.
A gene therapy that has been successful in treating an inherited, blinding eye disease in dogs is now ready for clinical trials in humans with the rare condition retinitis pigmentosa. The therapy introduces a normal copy of the CNGB1 gene, halting vision loss and potentially benefiting around 2 million people affected worldwide. Retinitis pigmentosa affects approximately 2 million people worldwide, with 100,000 cases in the US alone. The therapy has been successfully trialed in dogs and works by rescuing normal function in rod cells, preserving cone function, and preserving retinal structure by stopping photoreceptor degeneration.
A gene therapy that successfully treated dogs with an inherited eye disease is ready for clinical development in humans with retinitis pigmentosa, a rare genetic disease that causes vision loss. The therapy uses an adeno-associated virus vector to deliver a normal copy of the CNGB1 gene under control of a novel gene promoter, which ensures that the CNGB1 introduced by the therapeutic is only active in the target cell–the rod photoreceptor. The therapy rescues normal function in rod cells, halts the accumulation of toxic amounts of cyclic guanosine monophosphate, preserves cone function, and preserves retinal structure.
Engineered induced pluripotent stem cells (iPSCs) have been successfully transplanted into rodents with retinal degeneration and ALS, leading to the protection of cells in the eyes that support vision and the spinal cord cells that control movement. The iPSCs were engineered to produce glial cell line-derived neurotrophic factor (GDNF), which helps sustain diseased neurons. The cells were safe and did not cause tumors or other problems when transplanted into the animals for several months. This research marks an important first step toward achieving more personalized therapies for people with these debilitating conditions that currently have no cures.
Researchers in China have used CRISPR gene editing to correct a mutation that leads to retinitis pigmentosa in mice, leading to the mice regaining their sight and retaining it well into old age. The study team hopes this promising new method could soon be used to similarly restore people’s vision in years to come. Retinitis pigmentosa is one of the most common causes of blindness, affecting one in every 4,000 people.
Researchers in China have successfully restored the vision of mice with retinitis pigmentosa, a major cause of blindness in humans, using a new, highly versatile form of CRISPR-based genome editing called PESpRY. The system can be programmed to correct many different types of genetic mutation, regardless of where they occur within the genome. The study provides substantial evidence for the in vivo applicability of this new genome-editing strategy and its potential in diverse research and therapeutic contexts, in particular for inherited retinal diseases such as retinitis pigmentosa.