A 12-month-old boy in Portugal experienced a temporary fishy odor after eating fish, diagnosed with a rare, often underdiagnosed genetic metabolic disorder called trimethylaminuria, which improved as his metabolism matured and enzyme function improved, allowing him to eat fish normally by age three.
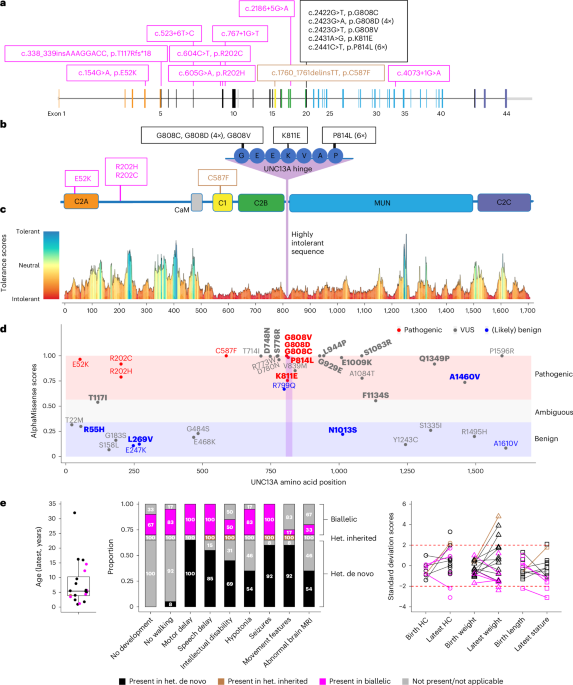
Pathogenic variants in UNC13A impair synaptic function, causing a neurodevelopmental syndrome with diverse clinical features, including profound developmental delay, seizures, and movement disorders, through mechanisms affecting protein expression and synaptic plasticity.
EMBL scientists developed SDR-seq, a new single-cell DNA-RNA sequencing tool that simultaneously studies both genetic variations and gene expression, especially in non-coding regions linked to diseases, enabling faster and more accurate disease connection discoveries.
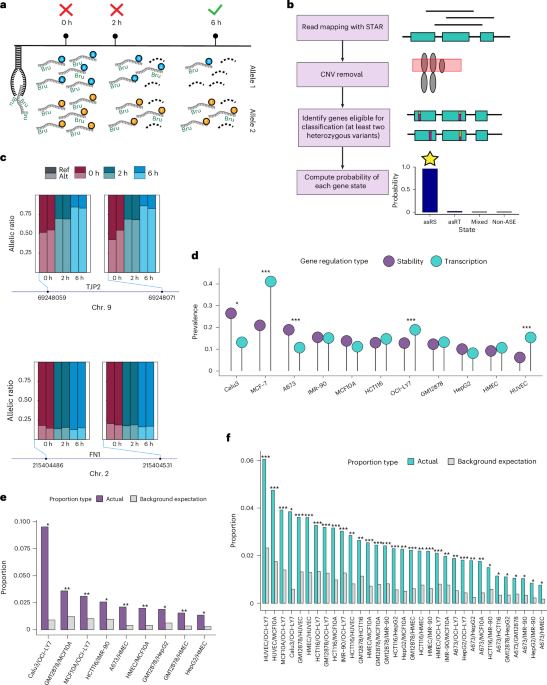
This study investigates how genetic variants affecting RNA stability influence complex traits and disease risk, utilizing extensive RNA sequencing data, eQTLs, and genome editing to identify functional variants and their roles in gene regulation and disease susceptibility.
A new statistical method has been developed to identify parent-of-origin effects in genes without needing parental data, revealing that certain genes can have opposite effects depending on whether they are inherited from the mother or father, with implications for understanding genetic influences on traits and diseases.
A new study suggests there may be millions of undiscovered genetic variants within the human species, challenging the long-held belief about our past and the extent of genetic variability. The "All of Us" research program is analyzing a large number of genomes and has already found over 275 million new genetic markers, potentially reshaping our understanding of human history. However, a recent study argues that genetic codes may not be as influential as previously thought, emphasizing the impact of external factors on gene expression. This raises questions about the significance of genetic research and its implications for our understanding of human development and history.
The National Institute of Health (NIH) has uncovered over 275 million new genetic variants in a study of 245,000 Americans, with nearly 4 million linked to higher risks of cancer, diabetes, and heart disease. This data, part of the 'All of Us' project, aims to build one of the largest genetic databases in the world by including diverse populations, particularly those from minority backgrounds. The findings could lead to new drug targets and treatments for specific populations, and help refine tools for assessing disease risk.
A study analyzing the genetic code of a quarter of a million U.S. volunteers has uncovered over 275 million new genetic variants, with nearly 4 million potentially linked to disease risk. The "All of Us" study aims to address the lack of diversity in existing genomic datasets and hopes to eventually collect DNA and health data from 1 million people to better understand genetic influences on health and disease. The findings highlight the importance of genetic diversity in understanding disease risk and developing effective drugs and prevention strategies for diverse populations.
The National Institutes of Health's All of Us Research Program has identified over 275 million new genetic variants from nearly 250,000 participants, half of whom are of non-European genetic ancestry. This unprecedented discovery provides new insights into genetic influences on health and disease, particularly in underrepresented communities. The findings, detailed in Nature, offer potential for advancing precision medicine for all populations and addressing health disparities. The program aims to engage at least one million diverse participants to contribute data for future scientific discoveries.
A new study has identified five biological pathways regulated by a few genes that could potentially play a prominent role in coronary artery disease through their involvement in endothelial cell function, including the gene TLNRD1. These findings could lead to the development of novel therapies targeting endothelial cell dysfunction in coronary artery disease, which is the leading cause of death in the United States. The study used high-throughput molecular biology techniques and computational methods to identify major biological pathways and novel genes involved in endothelial cell function that could contribute to the risk of coronary artery disease. This approach could facilitate the discovery of novel biological pathways associated with other diseases as well.
A large genetic analysis of Chinese parents and their babies has revealed multiple links between maternal health and fetal development, including genetic variants associated with maternal weight gain, bile acid levels, and liver disease risk. The study, published in Nature, is one of the first to examine the genetic profiles of East Asians and has identified discrepancies in the effects of certain genetic variants on mothers and their babies. While the findings offer new insights, further research is needed to confirm the associations and explore potential causality.
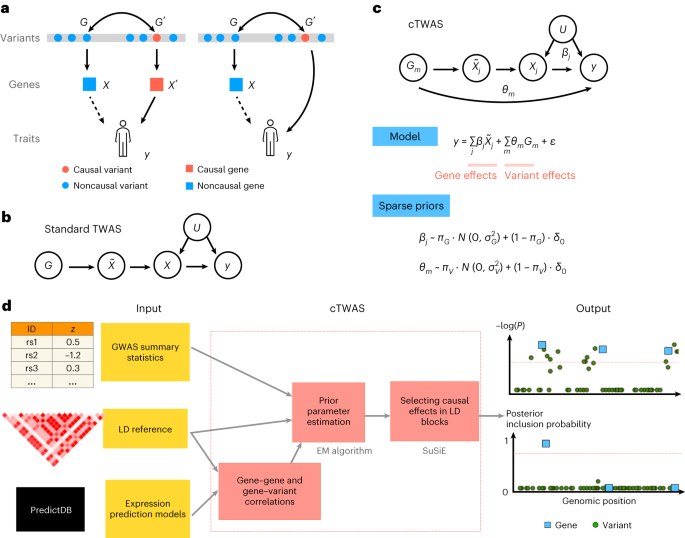
Researchers have proposed a new statistical framework called causal-TWAS (cTWAS) to address limitations in existing methods for transcriptome-wide association studies (TWAS). cTWAS aims to control for genetic confounders and improve the discovery of causal genes from genome-wide association studies (GWAS). Through simulations and real data applications, cTWAS demonstrated accurate parameter estimation, well-calibrated posterior inclusion probabilities (PIPs), and reduced false discoveries compared to standard TWAS, colocalization, and Mendelian randomization-based methods. In an application to GWAS of LDL cholesterol, cTWAS outperformed standard TWAS in distinguishing known LDL-related genes from nearby bystander genes, demonstrating its potential for reliable gene discovery in complex traits.
Ancient DNA analysis suggests that the higher risk of multiple sclerosis among people of northern European ancestry may be linked to genetic variants introduced by Bronze Age horseback-riding cattle herders who migrated into the region 5,000 years ago. These gene variants, which are known to increase the risk of multiple sclerosis, likely provided an advantage to the herders by protecting them from infections carried by their livestock. The findings shed light on the north-south divide in multiple sclerosis rates in Europe and may help explain the genetic basis of the disease.
Ancient DNA analysis reveals that the higher risk of multiple sclerosis among northern Europeans is linked to gene variants brought by the Yamnaya, a bronze age people who migrated into the region 5,000 years ago. These gene variants, which likely provided an advantage to the nomadic herders against infections carried by their livestock, are now associated with an increased risk of multiple sclerosis. The findings shed light on the genetic legacy of ancient migrations and offer a potential explanation for the north-south divide in multiple sclerosis prevalence in Europe.
Ancient DNA analysis reveals that the higher risk of multiple sclerosis among northern Europeans is linked to genetic variants brought by the Yamnaya, a Bronze Age people who migrated into the region 5,000 years ago. These gene variants, which are associated with increased MS risk, likely provided an advantage to the nomadic herders in fighting infections carried by their livestock. The findings shed light on the genetic legacy of ancient populations and offer a potential explanation for the north-south divide in MS prevalence in Europe.