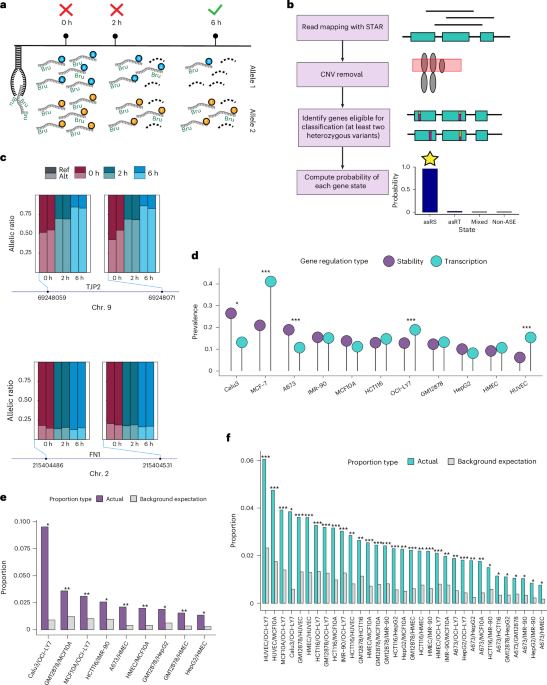
This study investigates how genetic variants affecting RNA stability influence complex traits and disease risk, utilizing extensive RNA sequencing data, eQTLs, and genome editing to identify functional variants and their roles in gene regulation and disease susceptibility.
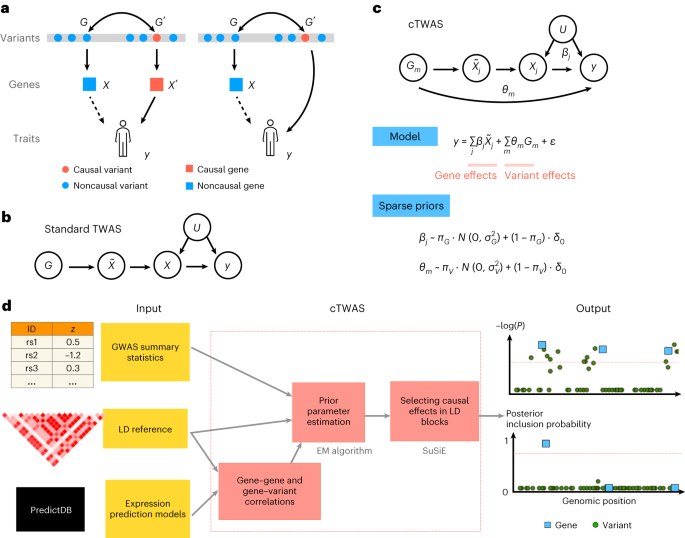
Researchers have proposed a new statistical framework called causal-TWAS (cTWAS) to address limitations in existing methods for transcriptome-wide association studies (TWAS). cTWAS aims to control for genetic confounders and improve the discovery of causal genes from genome-wide association studies (GWAS). Through simulations and real data applications, cTWAS demonstrated accurate parameter estimation, well-calibrated posterior inclusion probabilities (PIPs), and reduced false discoveries compared to standard TWAS, colocalization, and Mendelian randomization-based methods. In an application to GWAS of LDL cholesterol, cTWAS outperformed standard TWAS in distinguishing known LDL-related genes from nearby bystander genes, demonstrating its potential for reliable gene discovery in complex traits.
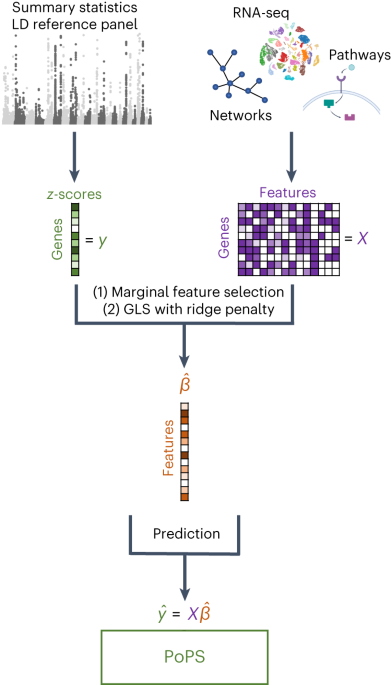
Researchers have developed a method called PoPS (Polygenic Prioritization of Gene Features) that leverages polygenic enrichments of gene features to predict genes underlying complex traits and diseases. The method utilizes various data sources, including genetic association studies, gene expression data, and functional annotations, to prioritize candidate genes. The researchers have made the processed gene features, visualizations, and code available on GitHub, as well as the PoPS results for multiple complex traits and diseases. This approach provides a valuable tool for understanding the genetic basis of complex traits and diseases.
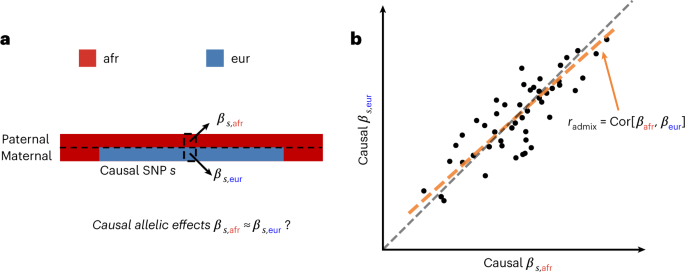
A study has found that the causal effects on complex traits are similar for common variants across segments of different continental ancestries within admixed individuals. The research used data from three large biobanks and found that the genetic architecture of complex traits is largely shared across populations, which has implications for the use of polygenic scores in diverse populations.