Scientists Identify Protein Key to Inflammation Control
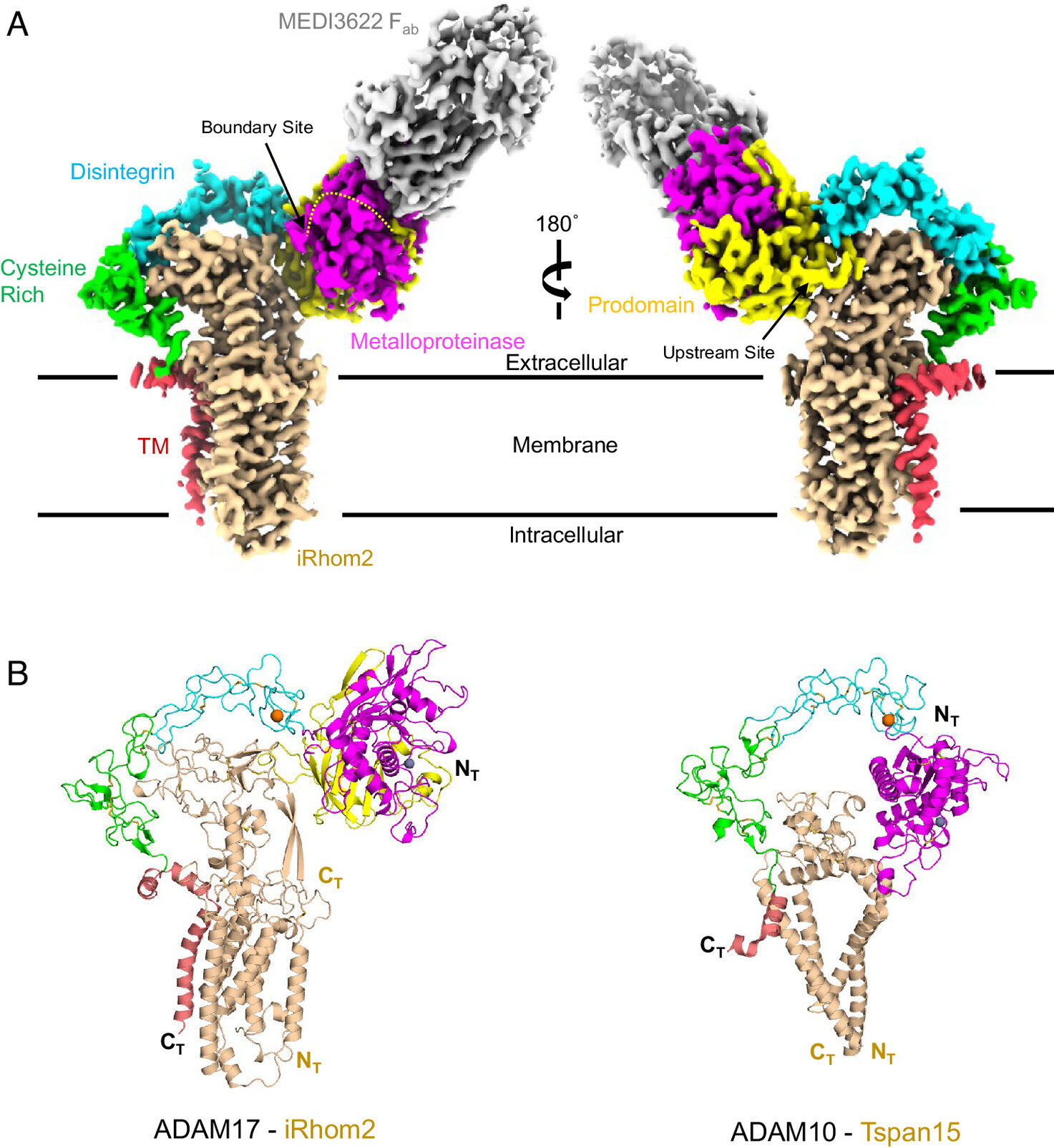
Researchers at the University of Cincinnati have used advanced cryogenic electron microscopy to visualize the atomic structure of the ADAM17 enzyme bound to its regulator protein iRhom2, revealing key features that could lead to targeted therapies for inflammatory diseases, cancer, and COVID-19. This breakthrough provides new insights into immune signaling and offers a foundation for developing more precise treatments.