A large-scale study using a novel computational method identified specific gut bacteria associated with autism spectrum disorder, highlighting the gut-brain connection and opening new avenues for research and potential therapies.
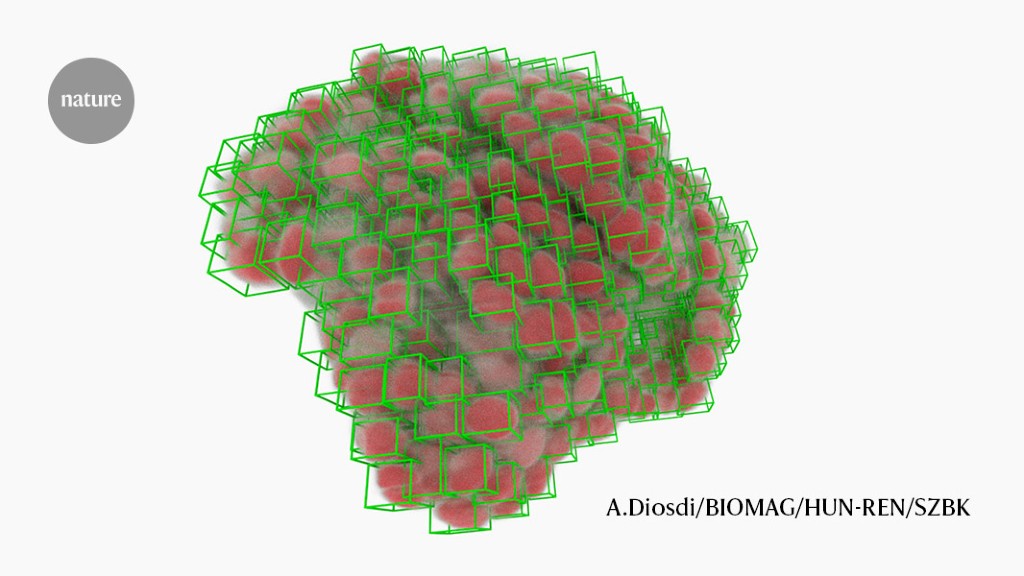
Computational biologists are harnessing the power of deep learning algorithms to improve the segmentation of cellular and subcellular features in biological imaging experiments. Algorithms such as U-Net have been transformative in identifying cell nuclei, while other approaches like StarDist and CellPose aim for a more holistic strategy by segmenting the entire cell. Training these algorithms requires large and diverse annotated datasets, and researchers are exploring strategies such as bulk annotation and human-in-the-loop approaches to streamline the process. While challenges remain, such as interoperability across imaging platforms and the analysis of 3D volumes, the field is making rapid progress and researchers are already exploring more advanced applications of these tools.
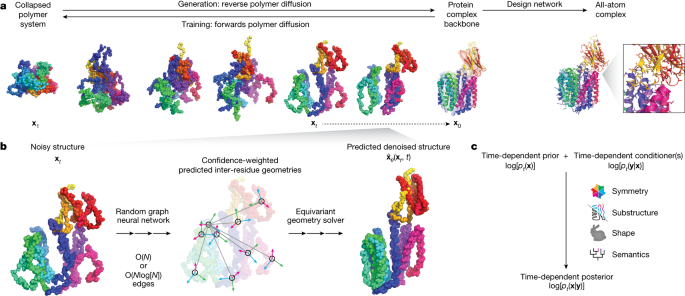
Researchers have developed Chroma, a generative model for proteins that can efficiently generate high-quality protein structures with diverse properties. Chroma combines diffusion models and graph neural networks to model the joint likelihood of sequences and three-dimensional structures of protein complexes. The model achieves quasi-linear computational scaling, allowing it to handle larger protein systems. Chroma also enables conditional sampling, allowing for the programmable generation of proteins based on desired properties such as symmetry, shape, protein class, and even textual input. This scalable generative model has the potential to significantly advance the design and construction of functional protein systems.
Ming Tommy Tang, a computational biologist, shares his unexpected career journey from a wet-lab biologist to a computational biologist. Tang emphasizes the importance of learning the command line, as it is essential for conducting bioinformatics analyses and working with high-performance computing clusters. He recommends resources such as online courses and books to learn the basics of the command line. Tang's dedication to helping other wet-lab biologists make the transition is evident through his blog and willingness to share his knowledge and experiences.
Stanford researchers have introduced Protpardelle, an all-atom diffusion model that co-designs protein structure and sequence. The model generates proteins of exceptional quality, diversity, and novelty by addressing the interplay between continuous and discrete protein structures. Protpardelle achieves high success rates in protein design, surpassing existing methodologies, and does so at a reduced computational cost. It demonstrates proficiency in generating diverse protein structures and has the ability to forge novel proteins beyond its training dataset. The model maintains chemical integrity and accurately captures sidechain behavior. Protpardelle's introduction marks a paradigm shift in protein design, with potential applications in biotechnology and pharmaceuticals.
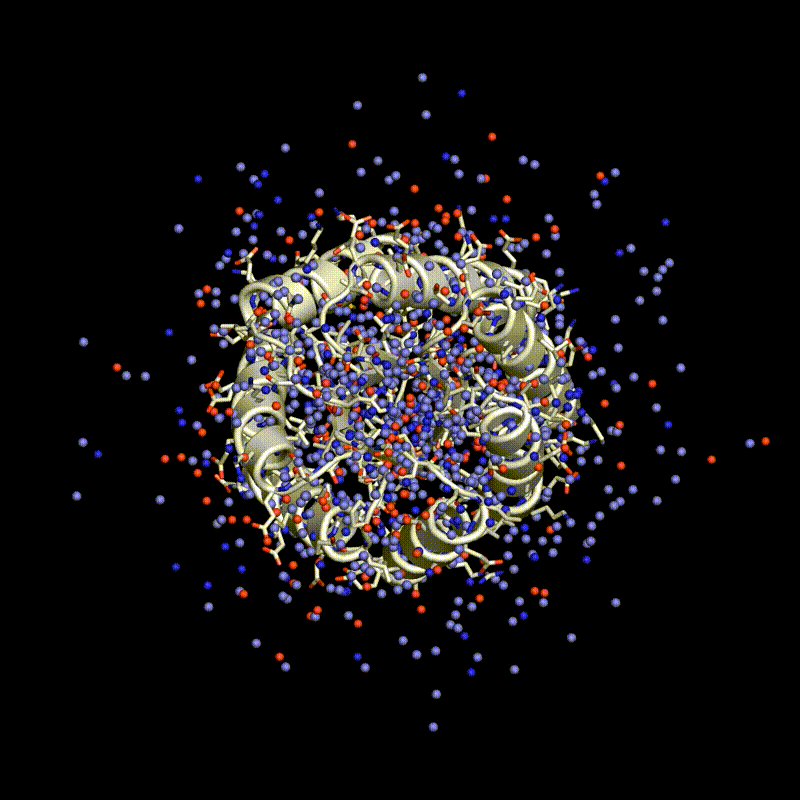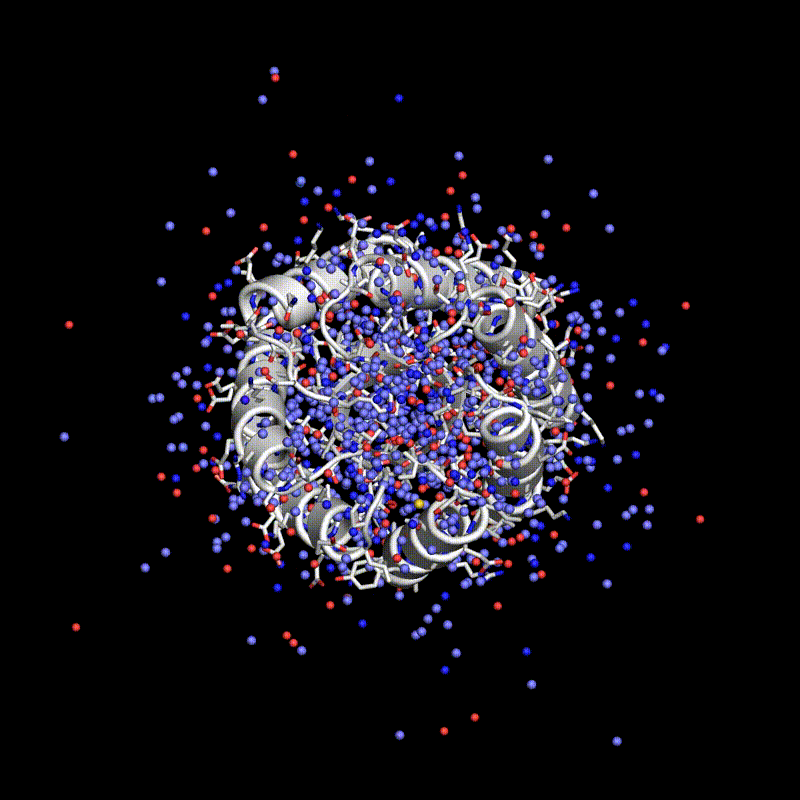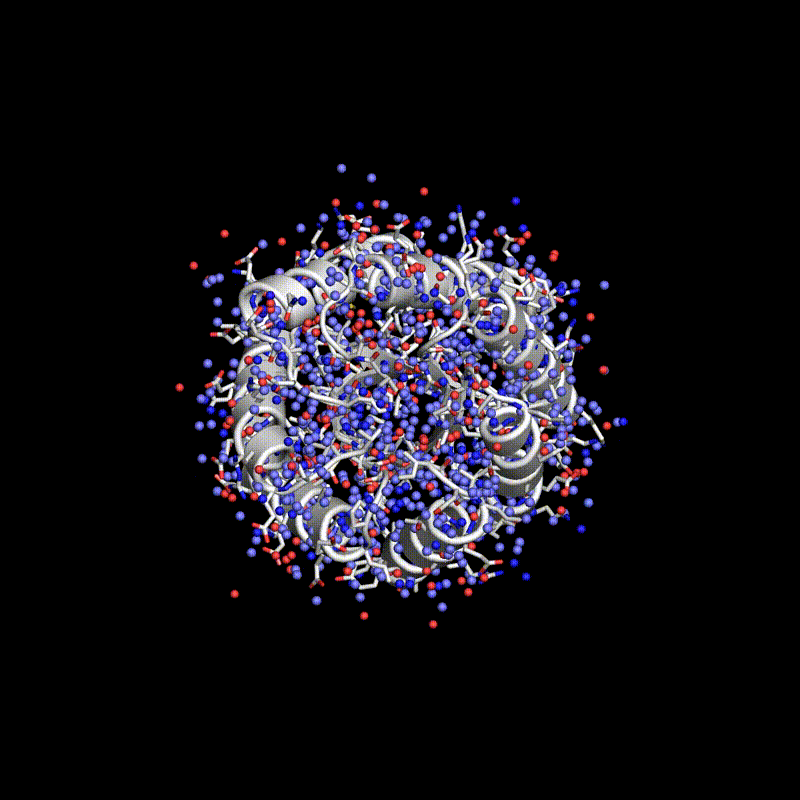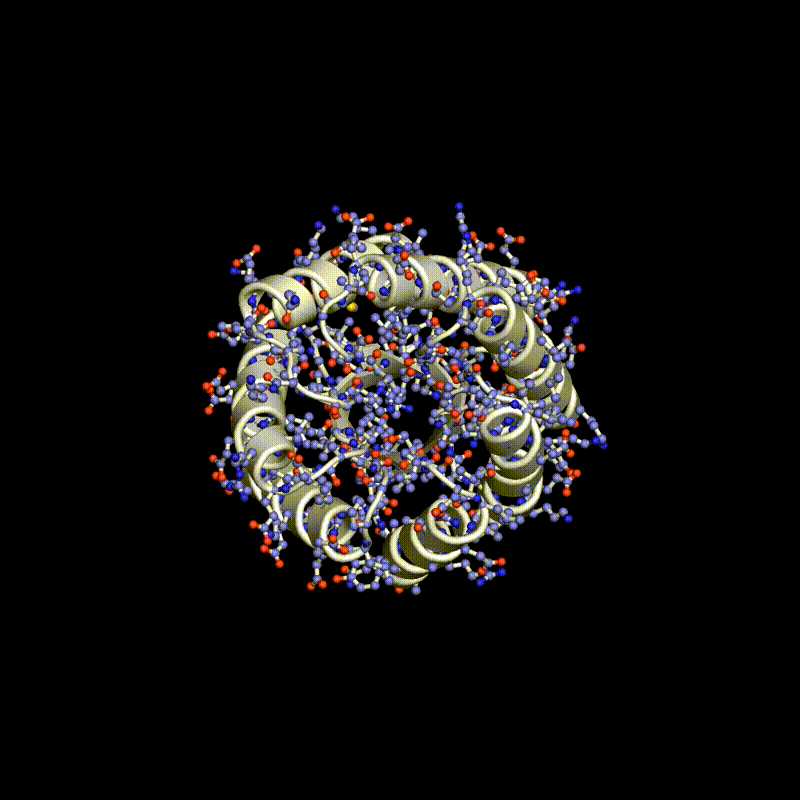
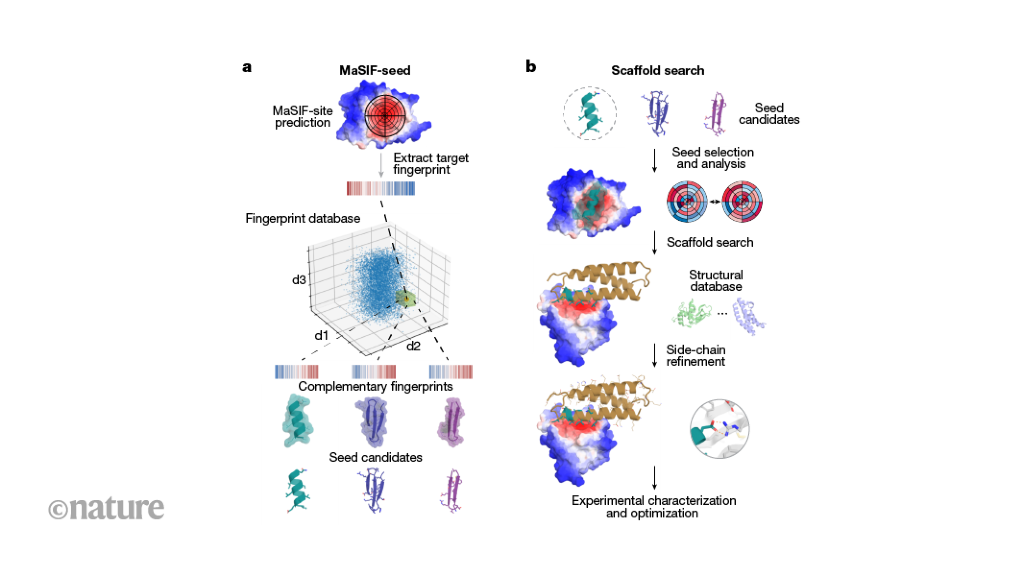
Researchers have used a computational approach to create synthetic proteins that can engage immunotherapeutic or viral targets with binding affinities comparable to those of naturally occurring proteins. The approach uses machine-learned fingerprints of protein-surface features to design protein interactions, which is a key challenge in the fields of basic and translational biology.